
Translate this page into:
Williams-Beuren Syndrome: A Rare Case from Western India
*Email: pankajkgadhia@gmail.com
Abstract
Williams-Beuren Syndrome (WBS) also known as Williams Syndrome (WS) is a rare multisystem genetic disorder having incidence of 1 in 20,000 to 50,000 live births. WS caused by deletion of 26 – 28 contiguous genes including elastin (ELN) on chromosome 7q11.23. It is characterized by congenital heart defects, skeletal and renal anomalies. We report herein two rare cases of WS (One male and one female) from Western India varying clinical presentation. The confirmation was carried out by cytogenetic analysis and FISH test.
Keywords
7q11.23
ELN
FISH
Heart Defects
Williams Beuren Syndrome

Introduction
Wiliams Syndrome (WS) (OMIM 194050) is a rare familial multisystem disorder occurring in 1 per 20,000 to 50,000 live births. It is characterized by congenital heart defects, facial dysmorphisms, mental retardation, skeletal and renal abnormalities and cognitive disorder1. WS is considered a segmental aneusomy due to a hemizygous deletion ~1.5 to 1.8 mb in the 7q11.23 region containing 26 to 28 genes and 2 mi RNA loci. Diagnosis of WS is made by clinical evaluation usually during infancy when they have distinctive facial appearance. The confirmation of diagnosis is carried out by Fluorescent In situ Hybridization (FISH) test. A clear genotype-phenotype correlation has been established in WS only for elastin gene which is responsible for the vascular and connective tissues abnormalities2. In the present communication, we present 2 cases with WS who have been referred to us with varying clinical features for cytogenetic study.
Case Presentation
Case – 1
A 02-year old girl child was born to non-consanguineous parents. The age of mother was 28 years and age of father was 30 years. The infant was born full term pregnancy by caesarean section. The birth weight was 2.62 Kg. The typical clinical features such as board forehead, facial dys-morphism, short nose, protruding lips were visible. The girl was friendly active child but she had a feeding problems.
Case – 2
A 08-year old boy was referred to us for cytogenetic analysis. He was born to non-consanguineous young couple. The maternal age was 26 years and paternal age was 30 years. There was a history of infertility. The boy was born full term normal delivery. The clinical examination clearly shows facial dysmorphism. The boy had broad forehead, upturned nose and lips were protruding. The boy had heart and feeding problems leading to failure of thrive, including gastrooesophageal reflux.
Methodology
The blood samples were collected from clinically confirmed WBS syndrome.
The whole blood culture was set up. FISH was carried out using LSI WS (Elastin gene – 7q11.23) probe (Vysis, USA). The slides were prepared and observed under a Zeiss Fluorescent microscope. The images were captured and processed. Figure 1 shows 46,XXish del (7q11.2) (ELN X1) (7q22 X2) ELN deletion compatible with WBS. While Figure 2 indicates 46,XY ish del(7q11.2) (ELN X1) (7q22) ELN deletion compatible with WBS.
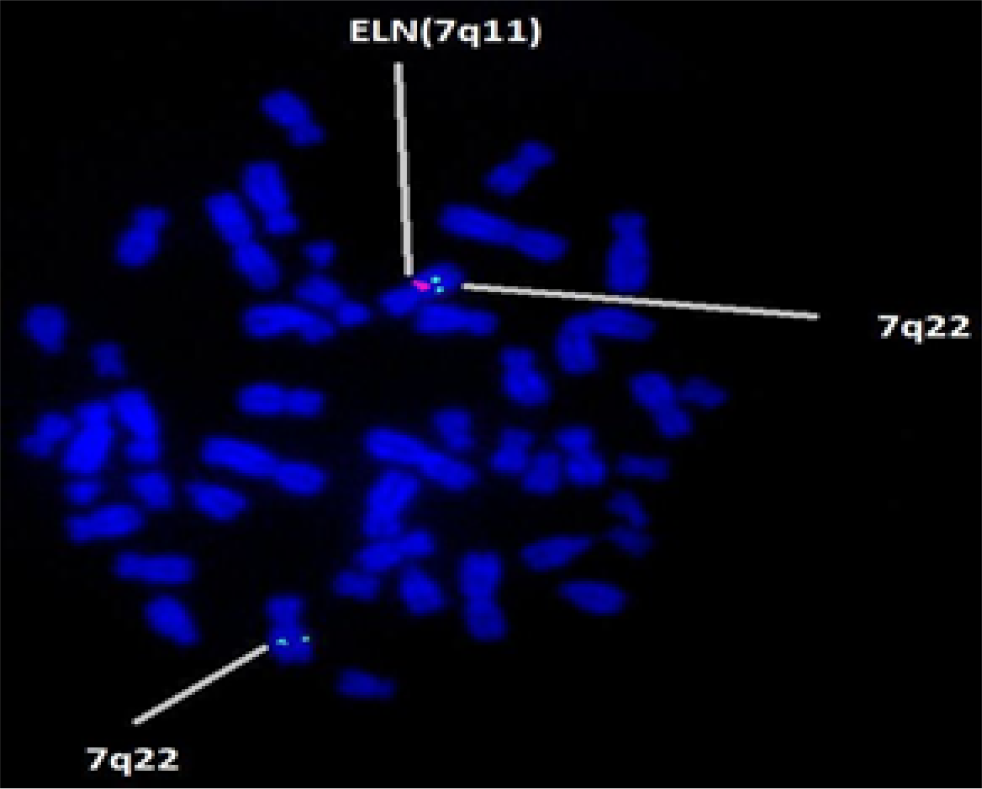
- FISH technique shows microdeletion of 7q11.23 containing the Elastin (ELN) gene locus in all metaphases.
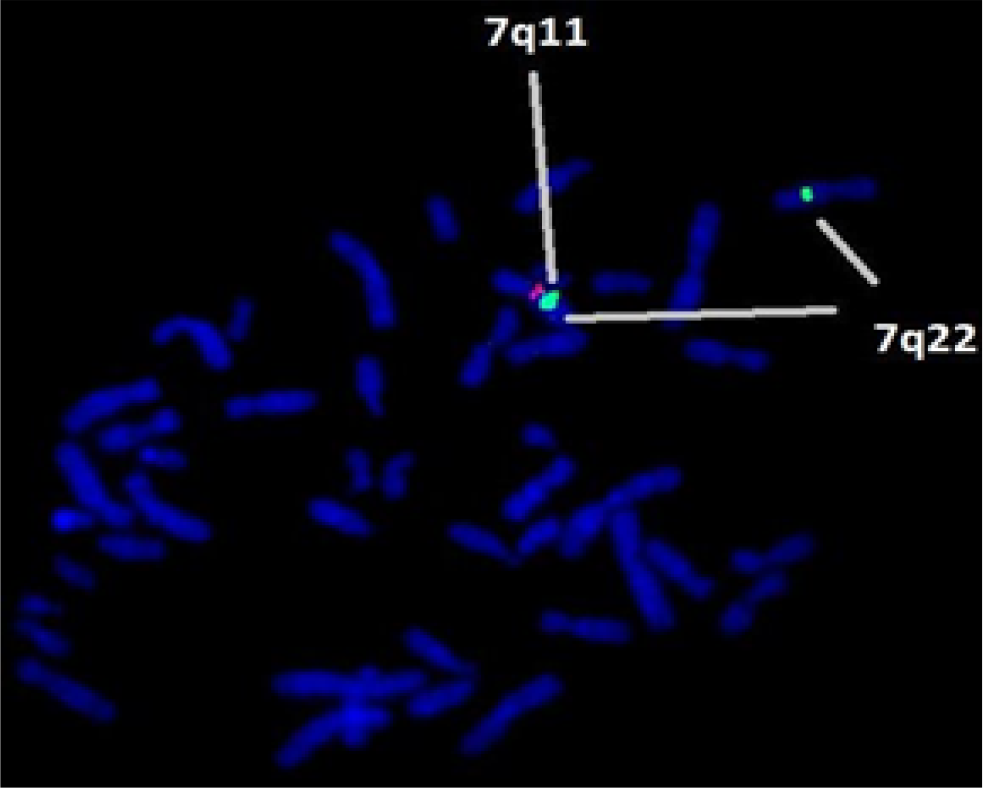
- FISH technique shows microdeletion of 7q11.23 containing the Elastin (ELN) gene locus in all metaphases.
Discussion
WS was first recognised as a distinct entity in 1961. It is present at birth and affects males and females equally. It can occur in all ethnic groups and has been reported throughout the world3. WS is caused by a well documented 1.6 mb hemizygous deletion of 26 – 28 genes on chromosome 7q11.23, a number of which play role for brain and behavioural features in WS4. Clinical features include cardiovascular anomalies, mainly supravalvulararotic stenosis (SVAS), peripheral pulmonary stenosis, distinctive facies, intellectual disability, unique personality and endocrine abnormalities. The most important cause of mortality in WS is SVAS which is present in 70%, of which 20 to 30% requires surgical corrections before 5 years of age5.
WS could account for 6% of all cases of mental retardation of genetic origin. In addition, many syndromes involving mental retardation are associated with behaviour and psychiatric traits6. The phenotype is characterized by short stature, craniofacial abnormalities. WS has frequent infantile hypercalcaemia leading to nephrocalcinosis7. WS arises due to genomic micro deletion at human chromosome 7q11.23 which is subjected to numerous genetic rearrangements.
In the present study the parents of both cases were cytogenetically normal and hence both the cases reported hereare sporadic events. Koolenet al.8 have also observed that the deletion of 7q11.23 is sporadic (De novo) in almost all cases. In our study, interestingly case -1 was not having any heart problems while case – 2 had Supra Valvular Arotic Stenosis (SVAS). Looking at the above facts, parents are advised for chromosomal analysis and FISH test along with child who is having abnormal clinical features. If the results are abnormal then they will be advised suitably.
Conclusion
According to primary clinical findings elfin face, congenital heart defects and neurocognitive defects WS is primarily diagnosed. The confirmation of 7q11.23 microdeletion must be supplemented with cytogenetic analysis and FISH techniques.
Acknowledgement
The authors would like to thank members of cytogenetic unit for their help.
References
- Williams Syndrome and related disorders. Ann Rev Genomics and Humn Genet. 2000;1:461-484.
- [CrossRef] [PubMed] [Google Scholar]
- Copy number variants at Williams-Beuren Syndrome 7q11.23 region. Humn Genet. 2010;128(1):3-26.
- [CrossRef] [PubMed] [Google Scholar]
- Sudden cardiac deaths under anesthesia in pediatric patient with Williams Syndrome: A case report and review of literature. Ann Cand Anthesia. 2005;13(1):44-48.
- [CrossRef] [PubMed] [Google Scholar]
- Cleft palate in Williams Syndrome. Ann Maxillofacial Surg. 2013;3(1):84-86.
- [CrossRef] [PubMed] [Google Scholar]
- Long term outcome of patients with cardiovascular abnormalities and Williams Syndrome. Am J Cardiol. 2010;105(6):874-878.
- [CrossRef] [PubMed] [Google Scholar]
- Williams Syndrome and psychosis: a case report. J Med Case Reports. 2014;4:49-51.
- [CrossRef] [PubMed] [Google Scholar]
- Idiopathic infantile hypercalcaemia in 5 month old girl. Prague Med Rep. 2011;112(2):124-31.
- [Google Scholar]
- Two families with sibling reoccurrence of the 17q21.3 microdeltion syndrome due to low grade mosacism. Eur J Humn Genet. 2012;20(7):729-33.
- [CrossRef] [PubMed] [Google Scholar]




